Die „dunkle Materie“ der Proteomik
Forschende am Max-Planck-Institut für Biochemie (MPIB) haben gemeinsam mit Kolleg*innen aus Madison, USA, herausgefunden, wie alternatives Spleißen und einzelne Nukleotidsubstitutionen auf Proteinebene übersetzt werden.
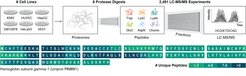
Die Shotgun-Proteomik wird zur Identifizierung einer Vielzahl von Proteinen in einer einzigen Probe eingesetzt. Aufgrund der begrenzten Anzahl identifizierter Peptidsequenzen sind die derzeitigen Techniken jedoch nur begrenzt in der Lage, zwischen den verschiedenen Produkten alternativen Spleißens und einzelnen Aminosäureaustauschen in Proteinen zu unterscheiden. Um diese „dunkle Materie“ der Proteomik anzugehen, haben Jürgen Cox, Gruppenleiter am MPIB und sein Team, zusammen mit Forschenden der University of Wisconsin-Madison und des Morgridge Institute of Research, eine neue Studie durchgeführt. Die Ergebnisse wurden in der Fachzeitschrift Nature Biotechnology veröffentlicht.


Alternatives Spleißen ist ein genetischer Mechanismus, der mehrere Proteinvarianten aus ein und demselben Gen erzeugen kann. Jedes Gen besteht aus mehreren „aktiven“ und „inaktiven“ Teilen. Während des gewöhnlichen Spleißens werden die inaktiven Genteile entfernt und die mRNA in der Reihenfolge gebildet, in der auch die aktiven Teile auf dem Ursprungsgen vorkommen. Während des alternativen Spleißens hingegen, werden die inaktiven Teile zwar ebenfalls entfernt, die aktiven Teile können aber in mehreren unterschiedlichen Reihenfolgen auf die mRNA übersetzt werden, wodurch eine vielfältige Population von Transkripten und Proteinen entsteht. Dieser Prozess kann die Genexpression regulieren, sich an gewebespezifische Bedürfnisse anpassen und eine funktionelle Vielfalt erzeugen, die für eine schnelle Evolution der Arten erforderlich ist. Bei Menschen können durch diesen Prozess mehr als 100.000 Proteine aus nur in etwa 20.000 Genen gebildet werden.
Die jüngsten technologischen Fortschritte in der Sequenzierung von Transkripten haben zur Identifizierung zahlreicher alternativer Spleißprodukte geführt. Die Frage, ob diese Produkte in funktionell unterschiedliche Proteine umgewandelt werden können, ist jedoch nicht direkt experimentell belegt. Dies liegt zum Teil an den Beschränkungen der derzeitigen Proteomtechniken, wie beispielsweise der Massenspektrometrie, die die am weitesten verbreitete Methode zur Sequenzierung des gesamten Proteoms ist. Die auf der Massenspektrometrie basierende Proteomik kann nur eine begrenzte Anzahl von Peptiden und damit nur einen kleinen Teil der insgesamt in einem Protein vorhandenen Aminosäuren identifizieren. Folglich werden Sequenzvariationen wie alternatives Spleißen und einzelne Aminosäuresubstitutionen in der Regel nicht erkannt, so dass es schwierig ist, ihren Beitrag zum Proteom genau zu bestimmen.
Umfassende Proteom Abdeckung
Ein bemerkenswerter Meilenstein auf dem Gebiet der Proteomik wurde kürzlich von einem internationalen Forschungsteam erreicht, an dem auch Jürgen Cox und sein Team am MPIB beteiligt waren. Das Team erreichte einen noch nie dagewesenen Abdeckungsgrad bei der Charakterisierung des gesamten menschlichen Proteoms, mit einer mittleren Sequenzabdeckung von 80 % für den Großteil der menschlichen Gene. Das Fachwissen des Teams für Computational Systems Biochemistry unter der Leitung von Jürgen Cox war hierbei entscheidend. Seine Gruppe trug durch die Analyse und Interpretation der enormen Menge von fünf Terabyte an experimentellen Daten zu der aktuellen Studie bei, was sich zuvor als erheblicher Engpass für die gesamte Initiative erwies. Diese Teamarbeit hat zusammen mit den anderen Instituten zu einem tieferen Verständnis des menschlichen Proteoms geführt, einschließlich bisher unbekannter Informationen über Aminosäuresubstitutionen und isoformspezifische Details.
Advanced Deep Proteomics
Obwohl es bereits ein Referenzproteom gibt und dieses ein wertvolles Modell für den Fortschritt der Biowissenschaften ist, besitzt in Wahrheit keine einzige Proteom-Probe seine Referenzeigenschaften. Zelllinien, Blutproben, sowie auch von Patient*innen stammendes Krebsgewebe enthalten jeweils einzigartige Aminosäureunterschiede, die von der Referenz abweichen. Um diese Unterschiede aufzudecken, hat das Team am MPIB innovative informatische Werkzeuge für die Erkennung von Aminosäurevarianten entwickelt, die sie unter dem Begriff Advanced Deep Proteomics zusammenfassen. Advanced Deep Proteomics ermöglicht es den Forscher*innen nun, genetische Unterschiede zwischen Proben zu analysieren und dadurch ein tieferes Verständnis dafür zu erlangen, wie Mutationen die Proteinexpression und -stabilität beeinflussen. Darüber hinaus schafft diese Studie einen Rahmen für die direkte Erforschung der allelspezifischen Expression, die für die Beantwortung grundlegender Fragen im Zusammenhang mit Krankheiten und der Weiterentwicklung personalisierter Medizin von entscheidender Bedeutung ist.
Durch die erfolgreiche Zusammenarbeit mit den Experten für alternatives Spleißen, Benjamin Blencowe von der Universität Toronto in Kanada und Robert Weatheritt vom Garvan Institute of Medical Research in Australien, hat das Team einen bahnbrechenden Meilenstein erreicht, indem es eine noch nie dagewesene Anzahl alternativer Spleißvorgänge identifiziert hat. Das Team konnte den ersten direkten Beweis erbringen, dass die meisten rahmenerhaltenden Isoformen auch übersetzt werden. Darüber hinaus führten sie eine umfassende Studie zu den Faktoren durch, die die Erkennung alternativer Spleißereignisse durch Proteomik beeinflussen. Die Ergebnisse der Studie werden künftig dazu beitragen, die Erkennung und Quantifizierung alternativer Spleißereignisse noch weiter zu optimieren.
[tb]