The “dark matter” of proteomics
Researchers at the Max Planck Institute of Biochemistry (MPIB), together with colleagues from Madison, USA, found, how alternative splicing and single nucleotide substitutions are translated into protein-level changes.
Shotgun proteomics is commonly employed for identifying a vast number of proteins within a single sample. Nonetheless, current techniques have limitations in distinguishing between the different products of alternative splicing and single amino acid substitutions due to the limited number of identified peptide sequences. To address this "dark matter" of proteomics, Jürgen Cox, group leader at the MPIB, and his team collaborated with researchers from the University of Wisconsin-Madison and the Morgridge Institute for Research to conduct a new study aimed at expanding the proteome coverage. The results have been published in the journal Nature Biotechnology.


Alternative splicing is a genetic mechanism that can generate multiple protein variants from the same gene. Each gene consists of several "active" and "inactive" parts. During ordinary splicing, the inactive parts of the gene are removed and the mRNA is formed in the same order as the active parts occur on the gene of origin. During alternative splicing, however, the inactive parts are also removed, but the active parts can be transcribed onto the mRNA in several different orders, resulting in a diverse population of transcripts and proteins. This process can regulate gene expression, adapt to tissue-specific needs, and generate functional diversity required for rapid species evolution. In humans, this process can create more than 100,000 proteins from only about 20,000 genes.
Recent technological advancements in transcript sequencing have led to the identification of numerous alternative splicing products. However, the question of whether these products can be translated into functionally distinct proteins lacks direct experimental evidence. This is partially due to the limitations of current proteomics techniques, such as mass spectrometry, which is currently the most widely used method for whole proteome sequencing. Mass spectrometry-based proteomics can only identify a limited number of peptides, and therefore only a small fraction of the total amino acids presents in a protein. Consequently, sequence variations, such as alternative splicing and single amino acid substitutions, are generally not detected, making it difficult to accurately determine their contribution to the proteome.
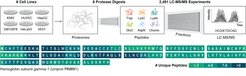
Deepest proteome coverage up to date
A remarkable milestone in the field of proteomics was recently achieved by an international team of researchers, which included Jürgen Cox and his team from the MPIB. The team accomplished an unprecedented level of coverage in characterising the entire human proteome, which led to a median sequence coverage of 80% across the majority of the human genes. The expertise of the Computational Systems Biochemistry team, led by Jürgen Cox, was essential. His group contributed by analysing and interpreting the massive five terabytes of experimental data, which was a significant bottleneck for the whole initiative in advance. This team effort, together with the other institutes, provided a more profound understanding of the human proteome, including previously unknown information on amino acid substitutions and isoform-specific details.
Advanced Deep Proteomics
Although there is a reference proteome, which has been a valuable model for advancing life science, in reality, no single proteomic sample possesses its reference property. For example, cell lines, blood samples, and patient-derived cancer tissue contain unique amino acid differences compared to the reference proteome. To address this issue, the team at the Max Planck Institute of Biochemistry developed innovative informatics tools for globally detecting amino acid variants. Resulting Advanced Deep Proteomics enables researchers to analyse genetic differences between samples and gain a deeper understanding of how mutations impact protein expression and stability. Additionally, this study creates a framework for directly exploring allele-specific expression, which is crucial for addressing fundamental questions related to disease and personalised medicine.
Through a fruitful collaboration with alternative splicing experts Benjamin Blencowe from the University of Toronto in Canada and Robert Weatheritt from the Garvan Institute of Medical Research in Australia, the team has achieved a groundbreaking milestone in identifying an unprecedented number of alternative splicing events. As a result, the team has provided the first direct evidence that most frame-preserving isoforms are translated. Moreover, the team conducted an extensive study of factors that affect the detection of alternative splicing events using proteomics. The findings of this study will assist in further optimising the detection and quantification of alternative splicing events.
[tb]