Molekulare Onkologie
Forschungsbericht (importiert) 2004 - Max-Planck-Institut für Biochemie
Onkogene und Tumorsupressoren
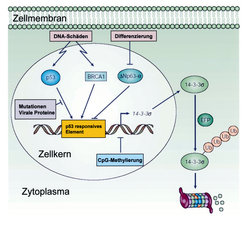
Zwei Klassen von Genen sind kausal an der Entstehung von Krebserkrankungen beteiligt: Tumorsuppressorgene und Onkogene. Erstere werden durch größere Deletionen, Punkt-Mutationen oder epigenetische Mechanismen inaktiviert. Onkogene dagegen werden durch genetische Veränderungen wie Amplifikationen und Punktmutationen aktiviert. Zwei prominente Vertreter dieser zwei Klassen von Genen sind das Tumorsuppressorgen p53 und das Onkogen c-MYC. Von beiden Genen ist bekannt, dass sie in mehr als 50% aller Krebserkrankungen verändert vorliegen. Die Forschung in unserer Gruppe beschäftigt sich mit verschiedenen Aspekten der Regulation und Funktion dieser Gene und ihrer Proteinprodukte. Sowohl das p53 als auch das c-MYC-Protein sind so genannte Transkriptionsfaktoren, die eine Vielzahl anderer Gene regulieren. Das p53 Protein wird durch DNA Schädigung, aber auch durch Onkogene wie c-MYC aktiviert. Daraufhin induziert das p53-Protein die Aktivität diverser Zielgene, welche sowohl eine Arretierung der Zellproliferation als auch die Auslösung des programmierten Zelltodes vermitteln können. p53 verhindert so, dass Zellen mit Defekten, die zu unkontrolliertem Tumorwachstum führen, sich teilen. Daher wird p53 auch als Wächter des Genoms bezeichnet. In Epithelzellen, von denen sich alle Karzinome ableiten, induziert p53 das 14-3-3sigma-Gen, welches ein Protein kodiert, das die Hemmung der Zellteilung vermittelt (Abb.1). Der Name 14-3-3 wurde aus der Position dieser Proteine nach einer zweidimensionalen Gelelektrophorese
abgeleitet.

14-3-3sigma: Ein p53 Effektor, der häufig epigenetisch inaktiviert wird
Die Untersuchung des 14-3-3sigma-Gens und seines Proteinproduktes stellt einen Schwerpunkt unserer Forschungen dar. So konnte gezeigt werden, dass das 14-3-3sigma-Gen in Tumorzellen nicht nur durch Mutation des p53- Gens, sondern auch durch eine chemische Modifikation der DNA im Bereich seines Promotors epigenetisch inaktiviert wird (zur Übersicht siehe [1]). Diese Modifikation wird als CpG-Methylierung bezeichnet und führt zur Abschaltung des 14-3-3sigma-Gens, ohne dass die genetische Information der DNA an sich verändert wird. Wir konnten zeigen, dass in den 41 untersuchten Prostatakarzinomen das 14-3-3sigma-Gen durch DNA-Methylierung abgestellt wurde [2].
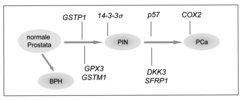
Die epigenetische Inaktivierung des 14-3-3sigma- Gens findet man ebenso im Basalzellkarzinom der Haut und im Brustkrebs. Die CpG-Methylierung des 14-3-3sigma-Gens kann in Zukunft von diagnostischer Bedeutung sein. Es ist nämlich möglich, Tumorzellen in Körperflüssigkeiten mithilfe einer CpG-Methylierungs-spezifischen PCR (Polymerase Kettenreaktion) nachzuweisen. Gegenüber dem Nachweis von Tumor-spezifischen Proteinen hat der Nachweis von CpG-Methylierungsereignissen den Vorteil, dass diese sehr stabil und selbst in geringen Mengen in Körperflüssigkeiten nachzuweisen sind. Basierend auf dieser Methode sollen in Zukunft diagnostische Tests zum Nachweis bestimmter Krebsarten entwickelt werden. Um weitere Gene zu identifizieren, die durch CpG-Methylierung im Prostatakarzinom inaktiviert werden, wurde eine genomweite Analyse aller CpG-methylierten Gene durchgeführt [3]. Hierzu wurden in Prostata-Karzinomzelllinien DNA-Methyltransferasen und Histon-Deacetylasen pharmakologisch inhibitert. Infolgedessen wurden Gene, die im Tumor durch DNA-Methylierung epigenetisch inaktiviert waren, wieder aktiv. Die wieder gebildeten mRNAs konnten anschließend durch Microarray-Analysen identifiziert werden. Nach Untersuchung von 50 ausgewählten Genen konnten 9 Gene mit potenziell tumorsuppressiven Funktionen identifiziert werden, die in 41 untersuchten Prostatakarzinomen in der Tat zu einem hohen Prozentsatz (zwischen 57 und 100%) epigenetisch inaktiviert vorlagen. Interessanterweise werden einige der Gene, die während der Seneszenz von Prostata-Epithelzellen induziert werden [4], wie zum Beispiel das DKK3- Gen, während der Tumorprogression durch CpG-Methylierung ihres Promoters inaktiviert (Abb. 2).
Um genetische Veränderungen im malignen Melanom zu identifizieren, haben wir die aus der SAGE-Methode abgeleitete digitale Karyotypisierung eingesetzt. SAGE (serielle Analyse der Genexpression) ist eine Methode zur Quantifizierung von kurzen DNA-Fragmenten, die stellvertretend für Boten-RNAs oder genomische DNA-Abschnitte sind. Kandidaten-Gene, die in identifizierten homozygoten Deletionen liegen, werden momentan untersucht.

Proteomics und 14-3-3sigma
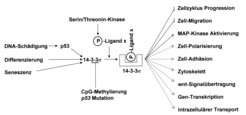
Nach Aktivierung des p53-Tumorsuppressorgenproduktes beeinflusst das induzierte 14-3-3sigma-Protein diverse zelluläre Prozesse durch Bindung an bestimmte Proteine. In einer Kollaboration mit der Arbeitsgruppe von Prof. John Yates III am Scripps Research Institute in San Diego/Kalifornien wurden 117 Proteine identifiziert, die mit dem 14-3-3sigma-Protein assoziiert sind [5]. Der verwendete, gezielte Proteomics-Ansatz beruht auf einer Methode (MudPIT= multidimensionale Protein Identifizierungs-Technologie), mit der komplexe Proteingemische aufgetrennt und anschließend die Bestandteile durch Massenspektrometrie bestimmt werden können. Die verschiedenen, bekannten Funktionen der mit 14-3-3sigma interagierenden Proteine deuten auf eine Vielzahl zellulärer Prozesse hin, die durch eine Induktion der 14-3-3sigma-Expression, vermittelt durch p53-Aktivierung, beeinflusst wird (Abb. 3). Das 14-3-3sigma-Gen ermöglicht somit dem p53-Tumorsuppressor-Protein, neben den durch Geninduktionen vermittelten Effekten eine Vielzahl von Prozessen durch phosphorylierungsabhängige Protein-Protein-Interaktionen zu regulieren. Die Rolle und Funktion dieser 14-3-3sigma/Liganden-Interaktionen wird momentan mit zellbiologischen Methoden untersucht.

Strukturelle Untersuchung des 14-3-3sigma-Proteins
Neben dem 14-3-3sigma-Protein gibt es noch sechs weitere 14-3-3-Proteine, die in menschlichen Zellen gebildet werden. Um zu verstehen, was diese Proteine unterscheidet, wurde in Zusammenarbeit mit der Arbeitsgruppe von Herrn Dr. Tad Holak (MPI für Biochemie) die Kristallstruktur des 14-3-3sigma-Proteins bestimmt (Abb. 4; [6])
![Struktur des 14-3-3sigma-Homodimers. Das Dimer bildet zwei Vertiefungen, in die jeweils eine Phosphopeptid-Kette des Liganden binden kann [6].](https://www.mpg.de/826916/original-1294357366.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6ODI2OTE2fQ%3D%3D--43153776dc79297b717033d85fad1eba839039c3 "Struktur des 14-3-3sigma-Homodimers. Das Dimer bildet zwei Vertiefungen, in die jeweils eine Phosphopeptid-Kette des Liganden binden kann [6].")

Dabei wurden strukturelle Unterschiede zwischen den von ihrer Aminosäuresequenz her hochkonservierten 14-3-3-Proteinen identifiziert, die wahrscheinlich determinieren, mit welchen Proteinen 14-3-3sigma interagiert. Zudem wurden Determinanten der Dimerisierungsspezifität identifiziert (Abb. 5). Derzeit wird mittels genetischer Methoden die funktionelle Relevanz der jeweiligen Aminosäuren, die für die Strukturunterschiede verantwortlich sind, untersucht.


Das Proto-Onkogen c-MYC
Das c-MYC-Gen wird in der Hälfte aller Krebsarten übermäßig gebildet und trägt wesentlich zur Tumorprogression bei (zur Übersicht siehe [7]). Da das c-MYC-Gen einen Knotenpunkt für die Vermittlung mitogener Signale darstellt, wird es nach Mutationen von Komponenten mitogener Signaltransduktionswege (wie z.B. SRC-Kinase, PDGF-Rezeptor, EGF-Rezeptor) induziert. In einigen Krebsformen ist das c-MYC-Gen zudem von Amplifikationen oder Translokationen betroffen. Das c-MYCProtein ist ein Helix-Loop-Helix-Leucin-Zipper Transkriptionsfaktor, der spezifisch an die so genannte E-box mit der DNA-Sequenz CACGTG bindet. Diese Sequenz befindet sich in der Promotor-Region c-MYC-regulierter Gene. Zudem unterdrückt das c-MYC-Protein antiproliferative Gene (wie den CDK-Inhibitor p21). Die Aktivierung von c-MYC ist ausreichend und notwendig für die Induktion der Zellzyklus-Progression in ruhenden Zellen. Obwohl diverse onkogene Funktionen des c-MYC-Proteins beschrieben worden sind, wie z.B. Induktion von Zellzyklusprogression, Immortalisierung, Transformation und Zellwachstum, war weitgehend unklar, welche Zielgene diese Effekte letztlich vermitteln. Um derartige Gene zu identifizieren, wurde mittels SAGE (Serielle Analyse der Genexpression; siehe oben) untersucht, welche Gene durch den Transkriptionsfaktor c-MYC induziert werden. Insgesamt wurden 216 Gene, die durch c-MYC induziert, und 260 durch c-MYC unterdrückte Gene identifiziert [8]. Dabei zeigte sich, dass c-MYC neben Genen der Zellzyklus-Regulation auch Gene induziert, die DNA-Reparatur, Neovaskularisierung, Protein-Translation/Abbau/Faltung, Glykolyse oder Apoptose vermitteln bzw. regulieren.
Derzeit versuchen wir, zusätzliche Funktionen und Regulationsmechanismen des c-MYC Proteins zu identifizieren, indem wir neue c-MYC-bindende Proteine mithilfe von Methoden, die auf Massenspektrometrie basieren (MudPIT), identifizieren. Diesen Ansatz hatten wir schon für die Suche nach 14-3-3sigma-Interaktionspartnern verwendet (s.o.). Die Ergebnisse dieser Untersuchungen deuten auf neue, für die Tumorbiologie relevante Funktionen des c-MYC-Proteins hin, die über seine Funktion als Transkriptionsfaktor hinausgehen.
c-MYC ist eines der wenigen Gene (bis auf virale Onkogene wie E1A oder SV40 T-Antigen), dessen Aktivierung für die Immortalisierung primärer Zellen ausreichend ist. Die so genannte Immortalisierung ist letztlich eine Aufhebung der in normalen Zellen vorhandenen Limitierung der Proliferationskapazität auf etwa 50 Zellteilungen (so genanntes Hayflick-Limit) und stellt ein Charakteristikum aller Tumorzellen dar. Wie c-MYC dies vermittelt, ist nur zum Teil verstanden. Die Erforschung der c-MYC-induzierten Immortalisierung ist ein Schwerpunkt unserer Untersuchungen und verspricht, einen Beitrag zum Verständnis der Alterung normaler Zellen zu leisten.