Zilien – die Antennen der Zelle
Forschungsbericht (importiert) 2014 - Max-Planck-Institut für Biochemie
Zilien – Motoren und Antennen für Zellen
Unser Körper besteht aus Milliarden spezialisierter Zellen, die sich bewegen oder miteinander kommunizieren müssen, um seine Funktion zu gewährleisten. Ein weithin bekanntes Beispiel für eine spezialisierte Zelle ist das Spermium, das möglichst schnell zur Eizelle schwimmen möchte und dafür ein bewegliches Flagellum, das sogenannte Zilium, besitzt. Bewegliche Zilien sind auch auf unbeweglichen Zellen, wie etwa in den Atemwegen, zu finden. Dort bewegen diese Zellen mit Hilfe von hunderten von Zilien Schleim und inhalierte Krankheitserreger weg von der Lunge, um Atemwegsinfektionen zu verhindern. Es ist deshalb nicht verwunderlich, dass Defekte im molekularen Aufbau von Zilien und Flagellen zu Unfruchtbarkeit oder zu schweren Atemwegserkrankungen führen.
Zilien sind aber nicht nur für die Bewegung von Zellen oder extrazellulärem Material verantwortlich, sondern spielen auch eine essenzielle Rolle bei der Verarbeitung von Sinneswahrnehmungen. Photorezeptorzellen in der Netzhaut des Auges benutzen Zilien zur Detektion von Licht, Nervenzellen in der Nase besitzen Zilien, die chemische Substanzen aus der Luft aufnehmen, und Zellen in den Gehörgängen registrieren Schallwellen, sobald diese auf spezialisierte Zilien auf der Zelloberfläche treffen und in Schwingung versetzen. Schäden innerhalb von Genen, die wichtig für den Aufbau und die Funktion von Zilien sind, führen deshalb oft zu Blindheit, Verlust des Geruchssinns und Taubheit.
Eine weitere Aufgabe von Zilien ist die Kommunikation zwischen Zellen. Dies ist besonders während der Embryonalentwicklung wichtig, bei der sich aus einer befruchteten Eizelle ein komplexer, multizellulärer Organismus entwickelt, in dem jede Zelle einen festgelegten Platz einnehmen muss. Sind Zilien schon im frühen Entwicklungsstadium in ihrer Funktion gestört, kann dies zu situs inversus, einer fehlerhaften Anordnung der inneren Organe, oder zu Polydactylie, einer falschen Anzahl von Fingern bzw. Zehen, führen.
Erwähnenswert ist auch die Verknüpfung der Ziliogenese mit dem Zellzyklus. Solange eine Zelle ein Zilium auf ihrer Oberfläche besitzt, wird die Zellteilung verhindert. Sobald aber ein extrazelluläres Signal die Zelle zur Teilung auffordert, wird das Zilium abgebaut und erst nach vollendeter Teilung wieder aufgebaut. Ziliäre Defekte gehen deshalb oft mit unkontrollierter Zellteilung einher, einem charakteristischen Merkmal von Krebszellen.
Alle hier aufgeführten Beispiele von ziliären Aufgaben machen deutlich, dass ein detailliertes Verständnis des Aufbaus und der Funktion dieser zellulären Antennen wichtig für das Verständnis schwerer Krankheiten des Menschen ist.
Wie werden Zilien gebaut?
 Chlamydomonas reinhardtii - eine einzellige Alge - unter dem Mikroskop. Das Bild zeigt die zwei langen Flagellen, die die Zelle zum Schwimmen und für die Fortpflanzung benutzt. (B) Schematischer Überblick über die wichtigsten Komponenten eines Ziliums, darunter der Basalkörper, das aus Mikrotubuli bestehende Grundgerüst, und die darüber liegende ziliäre Membran. Die IFT Maschinerie bindet die ziliären Bausteine, in diesem Fall das Protein Tubulin, und bringt sie von der Basis zur ziliären Spitze. (C) Das mit einem Elektronenmikroskop aufgenommene Bild zeigt einen Längsschnitt durch ein Zilium auf einer menschlichen Zelle.")
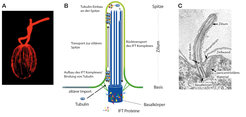
Zilien ragen aus Zellen heraus (Abb. 1), und dies stellt für die Zelle eine architektonische Herausforderung dar. Der Aufbau der Zilienstruktur beginnt von der Spitze her. Um die molekularen Bauteile zur Spitze zu bringen, besitzt die Zelle ein spezielles Transportsystem namens intraflagellar transport (IFT); dessen Maschinerie besteht aus mehr als zwanzig Proteinkomponenten, die einen großen Gesamtkomplex bilden und ziliäre Bestandteile von der Basis des Ziliums zur wachsenden Spitze befördern - ähnlich wie beim Bau eines Turmes. Dieser Mechanismus ist sehr alt und war höchstwahrscheinlich schon vor hunderten Millionen Jahren in den einfachen Vorfahren heutiger Zellen präsent.
Einer der ersten Schritte bei der Ziliogenese ist das Andocken des sogenannten Basalkörpers an die Zellmembran. Er besitzt eine definierte Anordnung von Röhren, sogenannte Mikrotubuli, die während des Wachsens des Ziliums verlängert werden und so das Grundgerüst formen. Baustein der Mikrotubuli ist das Protein Tubulin, und deshalb ist eine der wichtigsten Aufgaben der IFT-Maschinerie der Tubulin-Transport zur ziliären Spitze (Abb. 1B). Wie dieser Transport im Detail funktioniert, wird aktuell untersucht [1].
Wie wird an IFT Proteinen geforscht?
 Ein gereinigtes Protein nach Gelelektrophorese, angefärbt mit einem proteinspezifischen Farbstoff. (B) Zwei perfekte Kristalle eines IFT Proteins. (C) Ein Röntgenstrahlen-Streumuster auf einem Detektor, das durch den Beschuss eines Proteinkristalls mit Röntgenstrahlen entsteht.")

Um zu verstehen, wie die einzelnen IFT-Proteine zu einem großen IFT-Komplex zusammengebaut und wie der Transport ziliärer Bausteine ausgeführt werden kann, ist es sehr hilfreich zu wissen, wie die IFT-Proteine aussehen und wie sie miteinander interagieren. Dazu kann man die einzelnen Proteine reinigen, sie dann zu größeren Anordnungen zusammensetzen [2] und ihre dreidimensionale Struktur mittels Kristallographie erforschen (Abb. 2). Zuerst wird das Gen isoliert, das den jeweiligen Baustein kodiert, dann wird das Genprodukt in Bakterien oder Insektenzellen erzeugt und gereinigt (Abb. 2A). Das reine Protein wird danach kristallisiert (Abb. 2B), wobei sich viele Proteine in einer festgelegten Art und Weise aneinander anlagern. Derart kristalline Formen lenken Röntgenstrahlen ab und produzieren deshalb auf einem Detektor ein charakteristisches Muster (Abb. 2C). Diese sogenannte Röntgenkristallographie erlaubt die Rekonstruktion der Elektronendichte und somit der Position einzelner Atome des Proteins im dreidimensionalen Raum. Im Falle der IFT-Proteine konnte mithilfe dieser strukturbiologischen Methoden gezeigt werden, wie die Bausteine sich zu größeren Komplexen zusammenschließen und nachfolgend an Tubulin binden, um es zur ziliären Spitze zu transportieren. Mutationen, die diese Komplexbildung und somit den Tubulintransport verhindern, resultieren in gravierenden ziliären Defekten, die nicht kompatibel mit der Bildung eines lebensfähigen Organismus sind.
Wie kann man die molekularen Grundlagen von Ziliopathien bestimmen?
Dutzende Krankheiten des Menschen sind auf Defekte in der Ziliogenese zurückzuführen. Ausgelöst werden diese Ziliopathien teilweise durch Mutationen in Genen, die Bestandteile der ziliären Transportmaschinerie kodieren. Um Patienten effizient behandeln zu können, muss man verstehen, ob oder wie die jeweilige Mutation die Funktion des betroffenen Proteins verändert. Dazu kann man beispielsweise die mutierte Version des Proteins reinigen und mit dem ursprünglichen, Wildtyp-Protein vergleichen, zum Beispiel hinsichtlich der korrekten Faltung in die dreidimensionale Form oder hinsichtlich Interaktionen mit bekannten Bindungspartnern. Sobald die dreidimensionale Struktur des jeweiligen Proteins entschlüsselt worden ist, kann man zum Beispiel die Position einer eventuellen Veränderung im Molekül genau feststellen, was bereits wertvolle Informationen hinsichtlich der funktionellen Beeinträchtigung der Mutante liefert.
, die miteinander interagieren, um den korrekten Transport von Proteinen in ein Zilium zu gewährleisten. Die beiden Positionen, die häufig bei Patienten mit dem sogenannten Bardet-Biedl-Syndrom (BBS) mutiert sind, sind gekennzeichnet. Derartige Strukturuntersuchungen tragen zur Erklärung des Entstehens dieser Krankheit auf molekularer Ebene bei.")
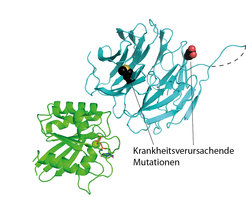
Die Kristallstruktur in Abb. 3 zeigt einen Proteinkomplex, bestehend aus zwei Proteinen, die am Aufbau eines Ziliums beteiligt sind [3]. Wird die Funktion dieser Proteine durch Mutationen beeinträchtigt, führt das beim Menschen zum sogenannten „Bardet-Biedl-Syndrom“ (BBS). Die Positionen zweier häufig auftretender Mutationen sind in der Abbildung markiert und eine davon liegt sehr nahe an derjenigen Stelle, an der sich die beiden Proteine aneinander anlagern. Dies deutet darauf hin, dass die mutationsbedingte Veränderung die Bindung der beiden Proteine stört, was nachfolgend mithilfe von biophysikalischen und biochemischen Methoden bestätigt werden konnte.
Für die meisten krankheitsverursachenden Mutationen sind solche strukturbiologischen Lösungsansätze jedoch noch nicht möglich und es zeichnet sich bereits jetzt ab, dass noch sehr viel Arbeit nötig sein wird, um die genauen Auslöser anderer Ziliopathien herauszufinden.
Literaturhinweise
Science 341, 1009-1012 (2013)