Protein folding
Proteins are chain-like molecules. In order to function properly, they must fold into complex three-dimensional shapes. The failure of proteins to fold properly has been linked to various diseases, including cancer, Huntington’s disease and Alzheimer’s disease. Understanding protein folding will aid the development of therapies that remove or prevent the formation of misfolded protein clumps.

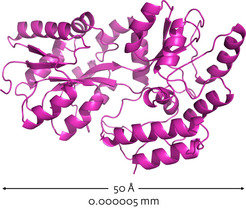
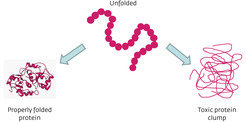
Proteins perform vital cellular functions; however, in order to work, these chain-like molecules must fold into complex, three-dimensional structures1 (Fig.1). The process is quality controlled by complex molecules within the cell2,3; however, it can go awry, yielding misfolded proteins that clump into toxic aggregates (Fig. 2). Many age-related diseases, such as Alzheimer’s disease, are thought to be caused by the build-up of these aggregates4,5. A co-ordinated network of interdisciplinary research is developing therapies that aim to remove or prevent the build-up of these misfolded proteins. If successful, their potential to improve quality of life is immense.
PROTEIN ORIGAMI
Proteins are the workhorses of the cell: they are essential molecules that fulfil vital and diverse cellular functions, such as catalysing chemical reactions, providing structural support, and mediating cell signalling and development. Human cells contain thousands of proteins, each consisting of a chain of 100–500 amino-acid building blocks. In order to work, however, these newly built proteins must first fold into a complex yet highly specific three-dimensional shape.

With hundreds of thousands of different possible structures for each protein, achieving the correct one is a tall order; yet, most proteins manage to perform this task quickly and efficiently. Larger proteins tackle the problem by folding different parts of the molecule separately. The process is driven by hydrophobic and hydrophilic properties of amino acids in different sections of the protein. Water-repelling amino acids interact with one other to build a hydrophobic core, whereas water-attracting amino acids settle on the exterior of the protein. This causes the protein chain to collapse into a globular structure that forms a stable, three-dimensional, biologically active protein.
QUALITY CONTROL
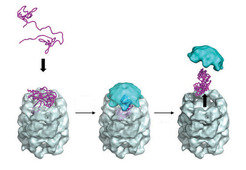
Given the complexity of the process, it is no surprise that the cell has evolved its own system of protein quality control. There are specialized proteins dubbed molecular chaperones, discovered in the late 1980s, that help prevent aberrant folding by shielding the sticky hydrophobic surfaces of unfolded or misfolded proteins. The members of one notable class of molecular chaperone, the chaperonins, take the form of tiny cylindrical cages with lids (Fig. 3). Unfolded proteins enter the cage and are protected as they fold, then released when the lid opens. Furthermore, each cell has a proteasome: a cylindrical complex that breaks up unneeded or misfolded proteins into small fragments, which can then be recycled to make new proteins.

Despite this sophisticated system of quality control, things can go wrong and misfolded proteins can accumulate. Aberrant folding can have dire consequences, as misfolded proteins might be unable to fulfil their biological roles. This can happen in two ways: the protein is either incorrectly constructed in the first place or loses its shape later in its lifetime. Examples of the former include mutations in the gene that encodes the tumour-suppressing protein p53, which can result in an incorrectly folded version of the p53 protein that cannot repair damaged — and potentially cancer-causing — snippets of DNA. Similarly, an inherited mutation in a key gene can trigger the formation of the misfolded protein that underlies cystic fibrosis.
As well as crippling protein function, aberrant folding can have other serious effects. Misfolded proteins can clump together to form insoluble aggregates that accumulate inside or around cells. It is thought that this build-up of cellular debris impairs cell functioning and is toxic to the cell. Cell death caused by the build-up of toxic aggregates is hypothesized to be a key feature of many age-related degenerative neurological disorders, including Huntington’s and Alzheimer’s diseases4,5.
THE RAVAGES OF TIME
Research suggests that our protein quality-control system functions less well with age, explaining why we might become more prone to the build-up of misfolded, potentially disease-causing proteins6. The brains of people with Alzheimer’s disease, for example, are riddled with hallmark misfolded protein aggregates called amyloid plaques. The plaques build up around nerve cells and, although it is not clear whether these aggregates are a cause or a consequence of the disease, it is thought that they contribute to cell death and memory loss.
The realization that protein misfolding might underlie a number of age-related diseases offers the opportunity to develop a generic therapy to address this group of economically costly and increasingly prevalent ailments. It has already been shown through a tissue-culture study that antibiotic treatment can boost the expression of several molecular chaperones and prevent the aggregation of mutant huntingtin protein, which causes Huntington’s disease7.
Although we understand protein folding at a basic level, its intricacies have yet to be revealed and key questions remain. How does our protein quality-control system work and why does it begin to fail with age? Why are misfolded proteins toxic to cells? What are they like structurally and how do they interact with the rest of the cell? How can we remove toxic protein aggregates or prevent their formation?
It is likely that the answers will hold major medical significance, as reflected by substantial research initiatives in the US, UK and Japan. Moreover, protein-folding studies, which were formerly merely of academic interest, are now the focus of intense applied therapeutic research.
GETTING IT TOGETHER
The challenge in understanding protein folding is in the pooling of relevant knowledge from different disciplines. Computer simulations and biophysical methods will help reveal the molecular features of protein aggregates; the toxicity of these aggregates will be assessed in tissue culture and animal models8. A technique called quantitative proteomics will help explain how misfolded proteins become toxic. Furthermore, research on the cellular quality-control system should shed light on how the cell controls its normal protein balance and any age-related changes. The goal is to help explain ageing and its associated diseases, and to develop relevant therapies.
Molecules that interfere directly with aggregate formation are already in development, as are strategies aiming to activate the defence mechanisms of the cell — chaperones and protein-degrading enzymes — to do the job9,10. Screening of large libraries of compounds will help identify new aggregate-busting and chaperone-activating molecules, and then drug-discovery researchers will assess any such molecules preclinically.
A detailed understanding of normal and aberrant protein folding is likely to require an integrated, interdisciplinary approach. This knowledge has the potential to improve our healthy lifespan and offer treatments for age-related disorders that are currently without cure.
Research at the Max Planck Institute of Biochemistry has provided insight into the mechanisms by which molecular chaperones can prevent the build-up of toxic protein aggregates in cells. This remarkable ability of chaperones could form the basis for the development of novel strategies in the fight against neurodegenerative diseases (Behrends, C. et al. Mol. Cell 23, 887–897, 2006).











