Research
Signal transduction and posttranslational modifications


Large-scale 'precision proteomics' based on mass spectrometry (MS) has emerged as an increasingly powerful and indispensable technology for studying PTMs because of its unbiased nature and the ability to locate the PTM with single amino acid resolution ( ![]() Chaudhary and Mann, Nat. Rev. Mol. Cell Biol., 2010,
Chaudhary and Mann, Nat. Rev. Mol. Cell Biol., 2010, ![]() Cox and Mann, Annu. Rev. Biochem., 2011,
Cox and Mann, Annu. Rev. Biochem., 2011, ![]() Humphrey, James and Mann, Trends in Endocrinology and Metabolism, 2015). Our signal transduction and PTM-proteomics team works on the global mapping and localization of posttranslational modifications (PTMs), in particular phosphorylation and ubiquitination in collaboration with our colleague Brenda Schulman ( Brenda Schulman’s lab) at our institute to better understand the signaling networks that regulate essentially all of the biology of cells and organisms in normal and disease states.
Humphrey, James and Mann, Trends in Endocrinology and Metabolism, 2015). Our signal transduction and PTM-proteomics team works on the global mapping and localization of posttranslational modifications (PTMs), in particular phosphorylation and ubiquitination in collaboration with our colleague Brenda Schulman ( Brenda Schulman’s lab) at our institute to better understand the signaling networks that regulate essentially all of the biology of cells and organisms in normal and disease states.
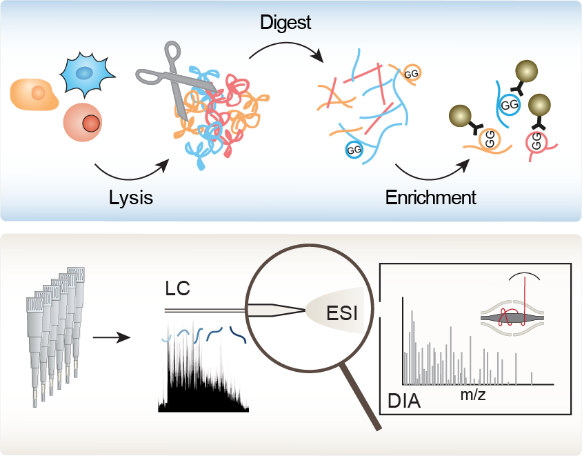
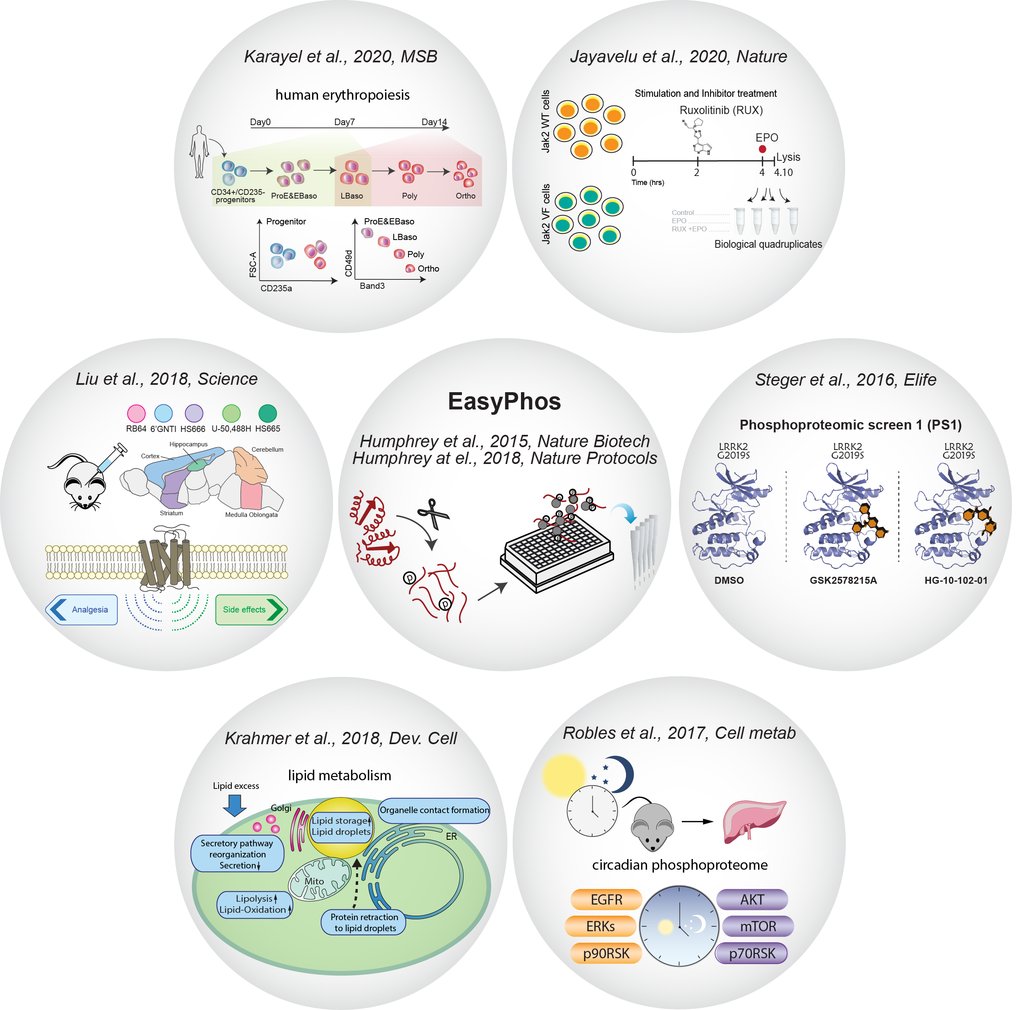 We have developed the EasyPhos technology (
We have developed the EasyPhos technology ( ![]() Humphrey et al., Nature Biotech., 2015,
Humphrey et al., Nature Biotech., 2015, ![]() Humphrey et al., Nature Protocols, 2018 ) that maximizes phosphoproteome coverage and reproducibility of phosphopeptide quantification in single liquid chromatography–MS (LC–MS) runs. We have employed this high-throughput platform to elucidate important signaling pathways in mouse brains (
Humphrey et al., Nature Protocols, 2018 ) that maximizes phosphoproteome coverage and reproducibility of phosphopeptide quantification in single liquid chromatography–MS (LC–MS) runs. We have employed this high-throughput platform to elucidate important signaling pathways in mouse brains ( ![]() Liu et al., Science 2018, islets isolated from diabetic (Leprdb/db) mice (
Liu et al., Science 2018, islets isolated from diabetic (Leprdb/db) mice ( ![]() Sacco et al., Cell Metab., 2019), lipid metabolism (
Sacco et al., Cell Metab., 2019), lipid metabolism ( ![]() Krahmer et al., Dev. Cell, 2018), circadian control (
Krahmer et al., Dev. Cell, 2018), circadian control ( ![]() Robles et al., Cell Metab., 2017) and human erythropoiesis (
Robles et al., Cell Metab., 2017) and human erythropoiesis ( ![]() Karayel et al., Mol. Syst. Biol., 2020). Funded by the Michael J. Fox Foundation for Parkinson’s research (
Karayel et al., Mol. Syst. Biol., 2020). Funded by the Michael J. Fox Foundation for Parkinson’s research ( ![]() www.michaeljfox.org), we identified a subset of Rab GTPases as bona fide substrates of the Parkinson's disease kinase LRRK2 (
www.michaeljfox.org), we identified a subset of Rab GTPases as bona fide substrates of the Parkinson's disease kinase LRRK2 ( ![]() Steger et al., Elife, 2016), and further investigated how LRRK2-mediated phosphorylation influences their function and downstream signaling pathways (
Steger et al., Elife, 2016), and further investigated how LRRK2-mediated phosphorylation influences their function and downstream signaling pathways ( ![]() Steger et al., Elife 2017). Recently we combined the phosphoproteome with validation approaches (using shRNA library) to create a functional phosphoproteome workflow for identification of novel signaling dependencies in cancer cells (
Steger et al., Elife 2017). Recently we combined the phosphoproteome with validation approaches (using shRNA library) to create a functional phosphoproteome workflow for identification of novel signaling dependencies in cancer cells ( ![]() Jayavelu et al., Nature, 2020).
Jayavelu et al., Nature, 2020).
We are also interested in the signal transduction pathways that govern immune signaling. Applying the Easy-Phos protocol, we recently achieved an in-depth insight into the phosphoproteome regulations by the cytokine TNF, thereby elucidating the kinetics and the localization of phosphorylation events and revealed upstream kinases mediating them ( Tanzer et al., Nat. Comm., 2021).
To extend the minimalistic approach of EasyPhos to antibody based post-translational modified peptide enrichment coupled to MS, we have developed a novel workflow called EasyAb. It allows rapid, sensitive, high-throughput, and cost-effective enrichment of modified peptides that requires antibody. Using this approach, we have explored phosphotyrosine signaling in cancer cell lines, patient samples and generated a long elusive in vivo map of leptin signaling in mouse brain.


We are currently optimizing a pipeline for the high-throughput analysis of phospho-samples. The combination of high-throughput sonication (COVARIS) and high-throughput phospho-enrichment (AssayMAP BRAVO) with short gradients on the Evosep enables us to measure thousands of phospho-samples within a short timeframe. In a systems biology we systematically perturb the phosphoproteome, which involves the processing and measurement of more than 1500 phospho-samples and their corresponding proteome. we apply the AssayMap Bravo workflow for studying the phosphoproteome at high throughput combined with trapped ion mobility spectrometry and an inherently fast qTOF (timsTof). Phosphopeptides possess a different distribution in the mass-to-charge and ion mobility plane, which requires the optimization of data-independent acquisition methods (dia-PASEF, see technological section) specifically for phosphoproteomics experiments. The dia-PASEF method was optimized with our tool Python package py_diAID (https://github.com/MannLabs/pydiaid) and quantified 35,000 phosphosites in only 21 minutes with 100 μg tryptic HeLa digest in the well-studied EGF pathway. This study demonstrates very deep coverage at high throughput and is one of the first studies of post-translational modifications with the PASEF principle (Skowronek et al., MCP, 2022).
 Moreover, we recently established a novel DIA-based ubiquitinome analysis workflow which obviates the need for high sample amounts and extensive fractionation of conventional workflows, thereby enabling an in-depth, system-wide analysis of dynamically regulated ubiquitinomes (
Moreover, we recently established a novel DIA-based ubiquitinome analysis workflow which obviates the need for high sample amounts and extensive fractionation of conventional workflows, thereby enabling an in-depth, system-wide analysis of dynamically regulated ubiquitinomes ( ![]() Hansen et al., Nat. Commun., 2021). This workflow increased the depth of analysis to more than 35.000 diGly peptides in single measurements. Its improved reproducibility enabled us to analyze for the first time the circadian cycle at the ubiquitinome level, unraveling hundreds of rhythmically oscillating ubiquitination sites, pointing to unexpected structural mechanisms and highlighting novel connections between metabolism and circadian regulation. We now apply our workflow to decipher signaling by ubiquitin and UBLs and elucidate how ubiquitin and UBLs switch the functions of their targets to regulate numerous biological processes. For this, we closely collaborate with the team of Brenda Schulman, the world’s leading experts in the field ( Brenda Schulman lab).
Hansen et al., Nat. Commun., 2021). This workflow increased the depth of analysis to more than 35.000 diGly peptides in single measurements. Its improved reproducibility enabled us to analyze for the first time the circadian cycle at the ubiquitinome level, unraveling hundreds of rhythmically oscillating ubiquitination sites, pointing to unexpected structural mechanisms and highlighting novel connections between metabolism and circadian regulation. We now apply our workflow to decipher signaling by ubiquitin and UBLs and elucidate how ubiquitin and UBLs switch the functions of their targets to regulate numerous biological processes. For this, we closely collaborate with the team of Brenda Schulman, the world’s leading experts in the field ( Brenda Schulman lab).


Furthermore, to provide fundamental information on the regulation and structure of biological systems, we investigate protein-protein interactions (PPIs). We have established workflows for affinity enrichment coupled to MS as a major tool for the mapping of PPIs in several organisms. For instance, we have successfully connected 5,400 proteins with more than 28,500 interactions in human systems without overexpression in ‘three quantitative dimensions’ ( Hein et al., Cell, 2015). Based on recent MS improvements in speed and sensitivity as well as the establishment of affinity enrichment coupled to MS ( Keilhauer et al., MCP, 2015, Hosp et al., MCP, 2015) we have developed a highly reproducible workflow for the large-scale identification of PPIs with very high completeness and quantitative accuracy on the Bruker timsTOF platform. In yeast, 4,159 pull-downs helped us to generate a highly structured network (yeast-interactome.org) that is similar to social networks between humans, revealing an average shortest distance of 4.2 interactions between proteins ( Michaelis et al.,Nature 2023 doi: 10.1038/s41586-023-06739-5). This technology was applied in a similar way in the ‘OpenCell’ effort by the Leonetti group at the Chan Zuckerberg Biohub to gain interaction and cellular localization information in human cell lines ( Cho et al., Science, 2022).