Clinical Proteomics

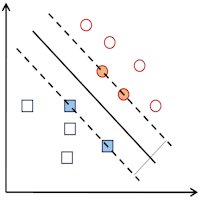
The analysis of proteomics data from patients requires special computational strategies. The problems that need to be addressed include questions like how to extract meaningful protein expression signatures from data with high individual variability, how to integrate the genomic background of the patients into the analysis of proteomics data and how to determine biomarkers and properly estimate their predictive power. We make use of machine learning algorithms like the support vector machine to classify patients and employ feature selection algorithms in order to extract predictive protein signatures. Different types of cross validation are used to assess the reliability of results.
Selected publications:
Geiger, T., Madden, S.F., Gallagher, W.M., Cox, J. and Mann, M. (2012). Proteomic portrait of human breast cancer progression identifies novel prognostic markers. Cancer Res 72, 2428-39.
Deeb, S.J., D’Souza, R.C.J., Cox, J., Schmidt-Supprian, M. and Mann, M. (2012). Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles. Mol Cell Proteomics 11(5), 77-89.
Deeb, S.J., Cox, J., Schmidt-Supprian, M. and Mann, M. (2014). N-linked glycosylation for in-depth cell surface proteomics of diffuse large B-cell lymphoma subtypes. Mol Cell Proteomics 13(1), 240-251.